Unit 06: An Introduction to Molecular Geometries
Contents
Unit 06: An Introduction to Molecular Geometries#

Authors:
Dr Antonia Mey
Dr Rafal Szabla
Email: antonia.mey@ed.ac.uk
Learning objectives:#
Read and manipulate molecular file formats:
xyzandpdbfilesVisualise molecules in a Jupyter notebook
Table of Contents#
Analysis of Molecular Geometries
1.1 Molecular File Formats
1.2 Measuring Structural Parameters
1.3 Tasks
Further reading for this topic#
Overview of different File formats in Chemistry: https://en.wikipedia.org/wiki/Chemical_file_format
Link to documentation:#
Tools we used in this session:
Tools that will allow you to read complex molecular file formats easily:
Jupyter Cheat Sheet
To run the currently highlighted cell and move focus to the next cell, hold ⇧ Shift and press ⏎ Enter;
To run the currently highlighted cell and keep focus in the same cell, hold ⇧ Ctrl and press ⏎ Enter;
To get help for a specific function, place the cursor within the function’s brackets, hold ⇧ Shift, and press ⇥ Tab;
Analysis of Molecular Geometries#
3.1 Molecular file formats#
Structural information about molecular systems can be obtained from various sources such as spectroscopy, crystallography, molecular dynamics or quantum chemical simulations. These data can be stored in various formats depending on their origin and intended applications. Apart from simple information about types of atoms and their positions in space, structural data may include information about bonding, protein or nucleic acid residues, atom types, properties (atomic numbers, charges), energies etc. In this section, you will learn how to visualize and manipulate structural data based on vector algebra. Most importantly, you will learn how to analyse complex files with molecular structures to understand and interpret various chemical phenomena.
Below you can find brief descriptions of the most popular file formats used to store information on molecular structure:
xyz files - contain atom names and their Cartesian coordinates in separate columns, in Angstroms. The first line defines the number of atoms, while the second line is a comment (e.g. energy, molecule name etc.)
pdb files - a more complex format with atomic Cartesian coordinates, most commonly used for biological systems. Contain information about atom types and residues.
Z-matrices - contain information about bonding parameters, valence angles and dihedral angles. Can be used for constructing simple molecules in ordinary text editors.
CIF files - crystallographic information files. Contain crystal structure and information about the periodic unit cell of the given material. This file format is the only format capable of describing periodic materials.
While there are many other file formats for representing molecular structures, we will primarily focus on the first three types. In particular, we will learn how to extract valuable information.
We will start with the simplest format, i.e. xyz files. Let’s start by considering a simple xyz file containing the structure of formamide. To view the contents of the formamide-min.xyz file in your directory, open the file, assign all its contents to a variable called molecule, and print it.
xyz files store x,y,z coordinates of a molecule and represent one of the simplest possible molecular file formats#
We can read the content of formamide-min.xyz using the open() command. This creates a file handle (called f, in this case), enabling us to interact with the file. Using the file handle, we can read all the content of the file calling the read() method.
f = open('data/formamide-min.xyz') # open the file
molecule = f.read() # read its content
f.close() #close the file
Understanding the content of an xyz file#
The first line gives information on how many atoms there are in the molecule
The second line provides a name for the molecule
The next six lines consists of 4 columns:
The first column is the name of the element,
The second, third, and forth give x,y, and z spatial coordinates, respectively.
print(molecule)
Let’s now repeat the same operations as above, this time loading a file in the pdb format.
f = open('data/formamide.pdb') # open the file
molecule_pdb = f.read() # read its content
f.close() #close the file
print(molecule_pdb)
Difference between pdb and xyz file formats#
The pdb file contains similar content to the xyz file, namely the coordiantes of the molecule
It also contains additional bonding information and has a very specific, more complex format
For visuatlisation purpose we will use the pdb format, for everything else xyz, due to its simplicity
NGLView is a tool for viewing the spatial coordinates of a molecule#
NGLView supports multiple file formats, but not natively xyz
You can use it to visualise trajectories of molecules (think molecular movie)
It is a handy tool to sanity check, e.g. bond lengths visually
You can use the
show_file()method to visualise the file
import nglview # Let's start by importing nglview
nglview.show_file('data/formamide.pdb')
Revisiting Dictionaries#
A dictionary is a table similar to lists and tuples, which uses keys instead of indices.
A key can be almost any Python object, e.g. string, number, tuple etc.
Use self-explanatory keys to keep Python code easy to understand.
Here follows an example of usage of a dictionary using information on the building blocks proteins: amino acids. There are 20 different naturally occurring amino acids. Let’s assume we have written a function that stores the number of different amino acids occurring in a single protein. The result can be conveniently stored in a dictionary:
amino_acids_in_protein = { "ALA" : 21, "GLY" : 14, "PRO" : 3, "CYS" : 2 }
The amino acid names are used as keys and the occurrence of each amino acid is used as the value.
⚠️ Remember: Dictionaries use curly brackets instead of square brackets (for lists) when initiated, but you can add a new item to a dictionary or query a value based on a key in the following way:
# Add a new pair
amino_acids_in_protein["TYR"] = 4
print(amino_acids_in_protein)
# Query the number of a given amino acid
print(f'There are {amino_acids_in_protein["ALA"]} "ALA" amino acids in the protein.')
The .keys() method returns all the keys in a dictionary. This is useful when you want to iterate over the dictionary in a loop:
for k in amino_acids_in_protein.keys():
print(f'{k} = {amino_acids_in_protein[k]}')
Parsing xyz files with Python#
The information provided by xyz files is limited to the positions of different atoms in 3-dimensional space. However, with some additional analysis we can learn more about the molecule itself, e.g.:
measuring bond lengths,
measuring angles,
calculating the numbers of atoms of a given elements,
calculating the molecular mass.
For this, we need to parse the file into separate lines. The file handle f enables us to do this via the the readlines() method. This allows us to create a list of all lines in the file.
f = open('data/formamide-min.xyz','r')
lines = f.readlines()
### remember to close the file
f.close()
print(lines)
The readlines() method creates a list of lines from a text file. Notice how:
each of them ending with the
\nEach line is stored as a string
The \n symbol is used to mark the of the line in text files on linux-based platforms.
For the majority of analyses, we will only need the lines containing atomic specification. Using list ranges, print only the lines containing atomic specifications:
print(lines[2:])
In order to extract the information on atom types and their positions, we need to further parse each line using the split() function (apart from the first two lines). This function generates a list of elements present in each line. Without any attribute, this method assumes that the elements are separated by spaces.
This is how you can separate a space-separated string of text (here, the fourth line of our formamide text file) into a list of elements:
list_of_elements = lines[3].split()
print(list_of_elements)
The opposite operation, i.e. joining a list into a single string with space-separated elements, can be carried out as follows:
some_text = ' '.join(list_of_elements)
print(some_text)
Tasks 4#
data/formamide-min.xyz file and count the number of atoms per atom type: how many Hydrogens, Carbons and Nitrogens are there? You will need to loop over the lines of the file, split the individual lines and then use the count() method for this. # Task 1: Test out the solution in this cell:
Click here to see solution to Task.
f = open('data/formamide-min.xyz', 'r')
lines = f.readlines()
f.close()
atoms=[]
for line in lines[2:]:
atomname = line.split()[0]
atoms.append(atomname)
print(f'List of all atoms in formamide: {atoms}')
print(f'Number of H atoms in this molecule = {atoms.count("H")}')
print(f'Number of N atoms in this molecule = {atoms.count("N")}')
print(f'Number of C atoms in this molecule = {atoms.count("C")}')
Create a dictionary
atom_masscontaining C, N, O and H atoms as keys and their atomic masses (in atomic units) as values.create a function taking two parameters: a dictionary containing atomic masses (such as the one created in the previous point), and the name of an xyz file. The function will iterate over the lines in the xyz file, and return the molecular mass of the molecule. Here you can exploit code written in the previous task!
Use the function to calculate the molecular mass of formamide, and on the file
data/colesterol.xyz. The latter should have a mass of 386.
# Task 2: Test out the solution in this cell:
Click here to see solution to Task.
# definition of the mass of each element
atom_mass = {'C':12, 'N':14, 'O':16, 'H':1}
def get_mass_of_molecule_from_file(atom_mass, file_name):
'''reads an xyz file and computes the atomic mass of the molecule in the file
Parameters:
-----------
atom_mass : dictionary
dictionary containing atomic masses
file_name : string
path to the xyz file to be read; only xyz is supported
Returns:
--------
mass_of_molecule : Integer
returns the molecular mass of the molecules in file_name
'''
# you could include a check if the file you are trying to read exists here
# reading file
f = open(file_name, 'r')
lines = f.readlines()
f.close()
atoms = []
for line in lines[2:]:
atom_name = line.split()[0]
atoms.append(atom_name)
no_C = atoms.count('C')
no_N = atoms.count('N')
no_H = atoms.count('H')
no_O = atoms.count('O')
mass_of_molecule=no_C*atom_mass['C']+no_N*atom_mass['N']+no_H*atom_mass['H']+no_O*atom_mass['O']
return mass_of_molecule
#call the function on formamide
mass_formamide = get_mass_of_molecule_from_file(atom_mass, "data/formamide-min.xyz")
print(mass_formamide)
#call the function on cholesterol
mass_colesterol = get_mass_of_molecule_from_file(atom_mass, "data/colesterol.xyz")
print(mass_colesterol)
3.2. Measuring Structural Parameters#
Calculating bond lengths, angles, or dihedrals is a typical task for a Chemist#
bonds, angles, and dihedrals are internal coordinates of a molecule (i.e. their value is independent on the specific position and orientation of the molecule). These can be used to represent molecular geometries in a z-matrix format.
bond lengths, angles, and dihedrals can be extracted from xyz files using vector algebra.
a bond can be seen as a vector between two points in space, A and B, where A and B are the coordinates of two atoms.
Bonds#
To compute the length of a bond, we need to know the length of the vector connecting two atoms A and B using this formula:
\(\vert\vert v\vert \vert\)=\(\sqrt{(x_B-x_A)^2+(y_B-y_A)^2+(z_B-z_A)^2}\)
Let’s look at an example:
A = [0.25, -0.1, 0.15]
B = [0.8, 0.65, 0.7]
What is the distance between A and B?
from math import sqrt
v=sqrt((B[0]-A[0])**2+(B[1]-A[1])**2+(B[2]-A[2])**2)
print(f'The distance between A and B is {v:.2f}')
Remember for real bonds you will also have a unit for the distance, typically Å!
Or using a built in function in Numpy#
np.linalg.norm is a quick way of working out the length of a vector.
Tasks 5#
Check that np.linalg.norm gives you the same answer.
Hint: you may need to convert the lists to NumPy arrays.
Click here to see solution to Task.
v=sqrt((B[0]-A[0])**2+(B[1]-A[1])**2+(B[2]-A[2])**2)
print(f'The distance between A and B is {v:.2f}')
dist = np.linalg.norm(np.array(A)-np.array(B))
print(f'The distance between A and B is {dist:.2f}')
Create a function called
bond_length. The function accepts as arguments a list of lines from an xyz file and two atomic indices, and returns the distance between the two atoms (in Å).Find the indices of the C and O atoms in the file
data/formamide-min.xyz(already parsed in thecoordinateslist in the cell below).Calculate the C=O bond length for the provided geometry of formamide (You should expect something around 1.2 Å).
# Test out the solution in this cell:
file = open('data/formamide-min.xyz','r')
lines=file.readlines()
file.close()
coordinates = lines[2:]
### your code here! ###
Click here to see solution to Task.
from math import sqrt
def bond_length(atom1_index,atom2_index,coordslist):
'''compute the bond length from two coordinates
Parameters:
-----------
atom1_index : integer
index of the first atom in the file
atom2 : integer
index of the second atom in the file
coordslist : list
list that contains strings of coordinates as parsed with an xyz file
Returns:
--------
bond_length : float
bond length in Angstrom
'''
#extract the atomic coordinates of two desired atoms
atom1 = coordslist[atom1_index].split()
atom2 = coordslist[atom2_index].split()
atom1_vec = [float(atom1[1]),float(atom1[2]),float(atom1[3])]
atom2_vec = [float(atom2[1]),float(atom2[2]),float(atom2[3])]
# define the vector |AB| connecting the two atoms
diff_vec = [atom2_vec[0]-atom1_vec[0],
atom2_vec[1]-atom1_vec[1],
atom2_vec[2]-atom1_vec[2]]
# calculate the length of the |AB| vector
bond_length = sqrt(diff_vec[0]**2+diff_vec[1]**2+diff_vec[2]**2)
return bond_length
print(f'The bond length of the C=O bond is : {bond_length(0,1,coordinates):.2f} Å.')
Angles#
Here is an example of different valence angles:
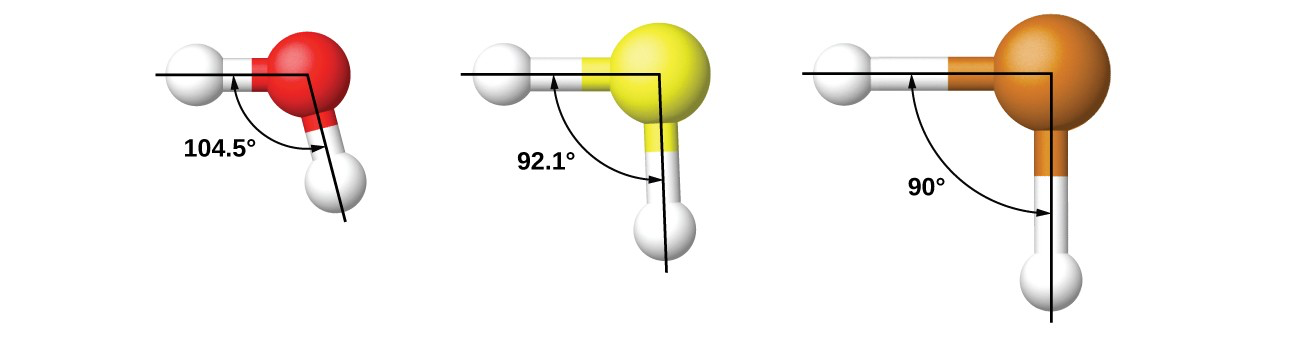
You can see, how an angle always includes 3 atoms. In the first example this is two hydrogen and an oxygen atoms. The angle can be computed from the bond between the first hydrogen and oxygen and the second hydrogen and the oxygen.
Here is an example of an angle in a water molecule, where vector H1, O, and H2 give the positions of the atoms in space.
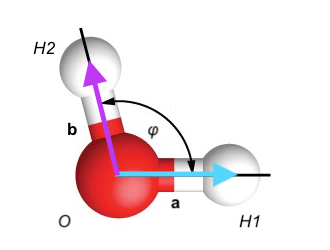
The bond length between H1 and O is given by the vector connecting these two atoms a in the image and can be computed using the above formula.
To determine the angle between two vectors you can use the scalar product:
where \(\mathbf{a}\) and \(\mathbf{b}\) are vectors, and \(\phi\) is the valence angle we are after. We need to solve the dot product according to the valence angle \(\phi\) by rearranging the above equation:
You can use the math library to get the arccos of an angle, e.g.: math.acos()
Measuring an angle in formamide#
Let’s try and measure the angle between some bonds in formamide. To measure the valence angle between the C-O and C-N bonds in formamide, you can reuse the coordinates we have loaded in the previous task.
We already have a way of working out \(\vert\vert\mathbf{a}\vert\vert \, \vert\vert\mathbf{b}\vert\vert\) with the function bond_length created in the previous task. What we still need to define, is a function that works out the vector for the C=O (e.g. \(\mathbf{a}\) in our example above) and C-N (e.g \(\mathbf{b}\) in our example) bonds and then we can define an angle. Let’s call this function get_vector_along_bond.
def get_vector_along_bond(atom1_index, atom2_index, coords):
'''retun the vector along a bond
Parameters:
-----------
atom1_index : integer
index of the first atom in the file
atom2 : integer
index of the second atom in the file
coordslist : list
list that contains strings of coordinates as parsed with an xyz file
Returns:
--------
bond_vector : list
bond vector contains the vector that describes the vector between
atom1_index and atom2_index
'''
x_coor=float(coords[atom1_index].split()[1])-float(coords[atom2_index].split()[1])
y_coor=float(coords[atom1_index].split()[2])-float(coords[atom2_index].split()[2])
z_coor=float(coords[atom1_index].split()[3])-float(coords[atom2_index].split()[3])
bond_vector=[x_coor,y_coor,z_coor]
return bond_vector
Tasks 6#
Create a function get_angle which takes in two bond lengths and two bond vectors to compute the angle between three atoms. Make use of the functions we have written so far. Do this for an example of the angle between O-C-N in formamide.
⚠️ The arccos is calculated with the function acos in math package, which returns an angle in radians.
# Task: Test out the solution in this cell:
from math import acos
import numpy as np
Click here to see solution to Task.
def get_angle(bond1_vector, bond2_vector, bond1_length, bond2_length):
'''Returns the angle between three atoms forming two bonds
Parameters:
-----------
bond1_vector : list
vector along bond1
bond2_vector : list
vector along bond2
bond1_length : float
length of bond1 in A
bond2_length : float
length of bond2 in A
Returns:
--------
angle : float
angle in radians
'''
theta = acos(np.dot(bond1_vector, bond2_vector)/(bond1_length*bond2_length))
return theta
CO_vector = get_vector_along_bond(0,1,coordinates)
CN_vector = get_vector_along_bond(0,3,coordinates)
CO_bond = bond_length(0,1,coordinates)
CN_bond = bond_length(0,3,coordinates)
theta = get_angle(CO_vector, CN_vector, CO_bond, CN_bond)
# theta is currently in radians. Let's convert it into degrees
theta_deg = np.degrees(theta)
print(f'The valence O-C-N angle amounts to {theta:.3f} radians or {theta_deg:.3f} degrees.')
There are many Python packages/libraries that can help you with tasks such as these#
… and many more
5. Molecular trajectories [Extra topic]#
Working with molecular trajectory files#
xyz files may contain more geometries written one after another.
This allows to store trajectories, e.g., information about the evolution of atomic coordinates of a given molecular system in time frame.
Trajectories can be generated through molecular dynamics simulations and subsequent structures correspond to different, consecutive time steps.
This format can be also used to store reaction paths, either optimized or obtained with simple interpolation between substrates and products.
Visualising a trajectory#
NGLView can be used to visualise a trajectory
This will require a pdb file with multiple frames in the same and xyz file can use multiple frames
Here is how you can visualise a trajectory of formamide stored in the file: data/formamide-traj.pdb
!pip install pytraj
import pytraj as pt
import nglview as nv
traj = pt.load('data/formamide-traj.pdb')
view = nv.show_pytraj(traj)
view
Every structure in the provided trajectory file corresponds to time step of 1 fs, and the entire trajectory is 100 fs long. In this particular file, every second line of each structure block contains the energy of the molecule (calculated with a semi-empirical density functional theory approach). Therefore, we can plot the time evolution of the energy of formamide.
Extra Tasks#
Read the trajectory file data/formamide-traj.xyz and extract the energy of the molecule at each time step using a for loop. Lines containing energy information start with the ' energy:' phrase, and can be located with a Python function. Append each energy measurement in the energies list, so that they appear in the same order as in the formamide-traj.xyz file. Store in the list timesteps the timestep associated to each energy measurement. Considering that the first geometry in the trajectory corresponds to the energy of the molecule after 1 fs of dynamics, and subsequent geometries were collected after 1 fs steps, you should get a list with 100 numbers from 1 to 100. The variable t contains information on the timestep being analysed. It is initialised as t=1, and needs to be incremented every time the script finds a line containing energy information.
# Task 1: Test out the solution in this cell:
# Hint:
traj = open(...
t = 1
energies = []
timesteps = []
for line in traj:
if line...:
energy = ...
...
Click here to see solution to Task.
file=open('data/formamide-traj.xyz','r')
traj=file.readlines()
file.close()
t=1
energies=[]
timesteps=[]
for line in traj:
if line.startswith(' energy:'):
energy=line.split()[1]
energies.append(float(energy))
timesteps.append(t)
t+=1
print(energies)
print(timesteps)
Energy information appears at regular intervals through the trajectory file. Above, we identified lines containing energy informations as those starting with ' energy:'. However, some data of interest appearing at regular intervals, may not be preceded by such a useful tag. Propose an alternative way of finding all of the lines containing the energy.
Hint: After parsing the file into a list of lines, use the formula giving the indices of all lines containing the energy. The energies can be extracted by iterating over the list of lines in the following way:
# Task 2: Test out the solution in this cell:
# Hint:
steps = 100
for n in range(steps):
lineindex = n*8+1
energy = traj[...
Click here to see solution to Task.
steps=100
file=open('data/formamide-traj.xyz','r')
traj=file.readlines()
file.close()
t=1
energies=[]
timesteps=[]
for n in range(steps):
lineindex=n*8+1
energy=traj[lineindex].split()[1]
energies.append(float(energy))
timesteps.append(t)
t+=1
print(energies)
print(timesteps)
Now we can use the two lists to plot the energy of the molecule in time frame. For this purpose you need to import the matplotlip.pylab module and use the plot(x, y) function. Use the timesteps as x and energies as y.
Click here to see solution to Task.
import matplotlib.pylab as plt
plt.plot(timesteps, energies)
plt.xlabel('timestep in [fs]')
plt.ylabel('Energy in kJ')
plt.show()